Definition
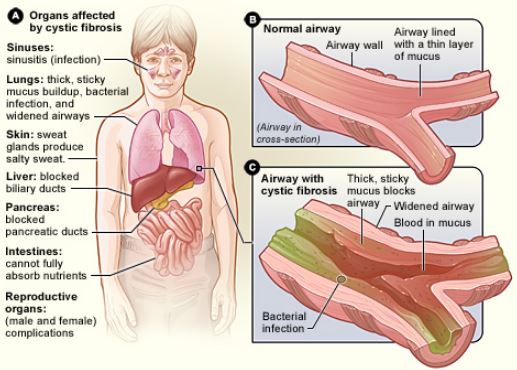
Cystic Fibrosis (CF) is a life-threatening genetic disorder primarily affecting the respiratory and digestive systems. It’s characterized by the production of thick, sticky mucus, which causes blockages in various organs, most notably the lungs and pancreas.
Causes
The genesis of Cystic Fibrosis lies in the mutations of the CFTR gene (Cystic Fibrosis Transmembrane Conductance Regulator). This gene plays a pivotal role in the production of mucus, sweat, and digestive juices. When the CFTR gene is defective, the resultant secretions become viscous and sticky. Two mutated genes, one inherited from each parent, are essential for an individual to manifest the disease. While carriers possess a single mutated CFTR gene, they typically don’t exhibit symptoms of CF. Yet, they hold a 25% chance of passing the condition onto their offspring if their partner is also a carrier.
Symptoms
The clinical presentation of CF varies between individuals, but the underpinning theme is the presence of thick mucus. The most commonly observed symptoms include:
- Respiratory Complications: Persistent coughing with thick mucus, wheezing, breathlessness, repeated lung infections, and nasal congestion.
- Digestive Issues: Due to mucus blockage in the pancreas, digestive enzymes are unable to reach the small intestine, leading to malnutrition and poor growth. This manifests as greasy, bulky stools, constipation, and intestinal blockages, especially in newborns.
- Sweat Abnormalities: Saltier-than-normal sweat is a distinctive sign, often leading to dehydration and an imbalance of electrolytes in the body.
- Reproductive System: Males with CF are usually infertile due to congenital absence of the vas deferens, while females may have difficulty conceiving due to thickened cervical mucus.
Treatment
Though CF is incurable, advancements in medical science have rendered its management more effective, enhancing the quality of life and increasing the lifespan of those afflicted:
- Medications: Antibiotics to combat lung infections, anti-inflammatory drugs to lessen swelling in the airways, and mucus-thinning drugs to help cough out the mucus more easily.
- Chest Physical Therapy: This involves pounding the chest and back to dislodge mucus from the lungs. Devices that aid in this process include vests with oscillating patches and handheld devices that one breathes out through.
- Pulmonary Rehabilitation: This encompasses exercise, breathing techniques, counseling, and nutritional advice aimed at improving lung function.
- Surgical Interventions: In advanced cases, a lung transplant might be necessary. Other procedures, like nasal polyp removal and feeding tube placement, might also be considered for alleviating symptoms and improving nourishment respectively.
In the context of CF, the importance of routine check-ups cannot be overstated. Monitoring facilitates early detection of complications, paving the way for timely interventions and fostering optimal patient outcomes.
Conclusion
In the vanguard of our battle against Cystic Fibrosis, rigorous scientific inquiry and patient-centered care hold the promise of brighter tomorrows. Our grasp of this intricate disorder strengthens with each passing day, bringing forth innovative strategies to alleviate suffering and extend life.